

Electrophoresis allows the proteins separated by SDS-PAGE to be transfered from the gel onto a membrane by electrophoretic transfer. In Part 3 of our Western blotting guide we detail the process of electroblotting and some of the considerations important to it’s success.
Overview
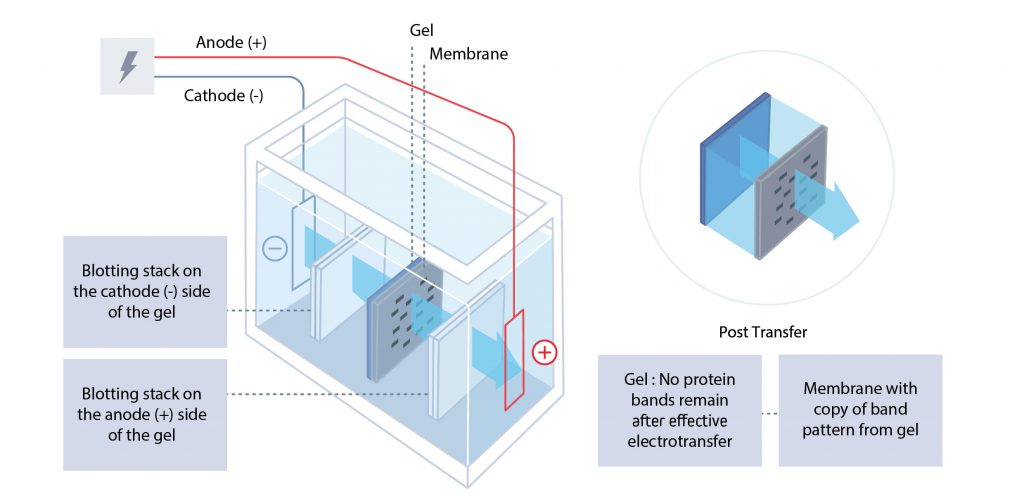
Following electrophoresis by SDS-PAGE, the separated proteins are electroblotted from the gel onto a membrane by electrophoretic transfer. The polyacrylamide gel and transfer membrane are placed in direct contact before being sandwiched between a negative electrode (cathode) and a positive electrode (anode).
The cathode is placed behind the gel, and the anode is placed behind the membrane. When voltage is applied, the proteins migrate from the gel to the membrane, where they are immobilized. The result is a mirror image of the gel pattern on the membrane. The blot (electroblotted membrane) is then blocked before immunodetection.
Types of Membranes
Membranes may be made from nitrocellulose or polyvinylidene difluoride (PVDF).
Nitrocellulose
Nitrocellulose membranes are non-hydrophobic and are believed to interact with proteins via noncovalent forces–electrostatic and hydrophobic properties.
Nitrocellulose is easily hydrated and offers a low background signal for most detection methods.
However, nitrocellulose membranes become brittle upon drying, making them difficult to handle if stripping and reprobing for alternative proteins are to be performed.
PVDF
PVDF membranes are chemically inert and more robust than their nitrocellulose counterparts, facilitating stripping and re-probing for alternative proteins. PVDF is hydrophobic and interacts with proteins through hydrophobic and dipole interactions. As such, they can form stronger interactions in comparison to nitrocellulose membranes, enabling them to potentially bind more protein (~150 µg cm ²).
However, greater membrane-protein affinity may increase the possibility of the membrane interacting with the primary and secondary antibodies, increasing the likelihood of background signal.
PVDF may auto-fluoresce, and low-fluorescence PVDF membranes must be used if fluorescent detection is to be performed to avoid background signal. Due to PVDF’s hydrophobic nature, membranes require “wetting” in methanol before blotting. This enables the buffer to permeate the matrix and allows the proteins to bind to the membrane.
Factors to Consider During the Transfer Step of Western Blotting
Pore Size
Membrane pore size may be a consideration if the protein of interest is particularly small or large.
Larger pores allow for a broader range of protein sizes but increase the risk of small proteins passing through the membrane during the transfer.
Whereas smaller pores provide a larger binding surface, increase binding capacity and reduce the chance of small proteins passing through the membrane. In conjunction with pore size, protein concentrations should be considered.
While high binding affinity between the membrane and proteins ensures sufficient protein binding for accurate analysis/detection, at high protein concentrations, this can result in protein-protein self-association rather than protein-membrane interactions and cause protein loss during the washing step.
Wash Steps
SDS can inhibit protein transfer to the membrane, and so the polyacrylamide gel may be washed with deionized water and then placed in the transfer buffer for 15–30 minutes to equilibrate to the buffer.
However, during this process, small proteins can migrate out of the gel, so careful consideration should be taken before performing a washing step. The sample is suspected of containing proteins with many hydrophobic amino acids, or high molecular weights, a transfer buffer with a low concentration of SDS (0.02 to 0.1%) may be used to prevent their precipitation.
Buffer pH
The transfer buffer maintains a pH above the protein’s isoelectric point, ensuring they retain a negative charge and migrate towards the anode.
A common transfer buffer uses the Towbin system (192 mM glycine, 25 mM Tris, 20% methanol). This low ionic strength buffer has a pH of 8.3, which is higher than the isoelectric point of most proteins.
As well as the salt type and concentration, the addition of acidic or basic solutions to adjust the pH affects the ion content and, consequently, the buffer’s conductivity.
High ion levels lead to increased running temperatures, which may result in inefficient transfer and an increase in the potential of undesirable artifacts.
Addition of Methanol
Methanol (MeOH) may be added to the transfer buffer to improve protein-membrane transfer efficiency. The addition of MeOH may cause the gel to shrink, increasing the gel density and retarding migration, particularly with high molecular weight proteins.
To prevent this, the gel is equilibrated in increasing volumes of MeOH until the required concentration is reached.
Analytical grade MeOH is recommended as lower grades may contain metallic impurities, damaging the apparatus, and should be avoided.
MeOH should be added just before use to prevent deterioration of the buffer or evaporation of the solvent.
Voltage
After the gel-membrane sandwich is assembled, the cassette is placed in a tank of buffer and voltage is applied to drive the transfer of proteins from gel to the membrane.
The voltage intensity and period that voltage is applied influence the efficiency of protein transfer and must be optimized for each system.
Low voltage over a short duration can result in incomplete transfer, whereas high voltage for an extended period will heat the system, leading to improper transfer.
High voltage may also result in small proteins passing through the membrane without binding. This can be observed by placing an additional membrane behind the first to capture these polypeptides.
Ideally, conditions should be optimized to ensure all sizes of proteins being analyzed transfer satisfactorily.
Electroblotting Methods
There are three different electroblotting methods: wet, semi-dry and dry. In all methods, the gel and the membrane are placed in direct contact, ensuring all air has been expelled between them.
| Method | Advantages | Disadvantages | |
|---|---|---|---|
| Wet Transfer | Gel-membrane is sandwiched between layers of filter paper and sponge and completely submerged in a tank of transfer buffer. | Highest quality blots in terms of transfer efficiency and distinctness of bands. Most suitable method for large proteins. | Generally has a shorter duration. However, high field options are available for a single gel transfer, which can significantly decrease the time taken (up to 30 minutes). Given the high voltage/current, this approach requires a cooling system to dissipate the heat generated. |
| Semi-Dry Transfer | Uses layers of filter paper soaked in a transfer buffer, removing the use of a tank. The membrane and filter papers are cut to be the same size, with no overhangs. This avoids producing a route that the current can use to bypass the gel entirely or create an inefficient or uneven transfer. | More cost efficient electrodes. | Gel drying and buffer depletion can occur if the transfer is performed for too long.
|
| Less complicated power packs. | This method produces lower quality blots, especially for larger proteins, but the time reduction is beneficial for many researchers. | ||
| The ability to blot several gels simultaneously. | |||
| Dry Transfer | Has a unique gel matrix that incorporates the buffer and eliminates the need for soaked filter papers and tanks. | Using dry transfer further reduces the time required, as little as 7 minutes, due to the short distance between the electrodes and no buffers are required. This method uses copper electrodes, which do not generate oxygen gas, a common cause of blot distortion in wet and semi-dry transfer. | As with semi-dry transfer, it will not produce as high-quality blots as the wet transfer method. |
Confirmation of Protein Transfer
Visual inspection of protein marker transfer allows the efficiency of protein transfer to be roughly assessed. However, temporary membrane staining is a more accurate method of determining protein transfer. Ponceau S (0.1% w/v Ponceau S, 5% v/v acetic acid in water) is a quick and easy stain compatible with both nitrocellulose and PVDF membranes.
Once stained, the proteins appear as red bands. The intensity of the bands indicates the amount of protein bound to the membrane and signifies the blot uniformity.
Bubbles, which may interfere with protein transfer, appear as blank circles.
Ponceau S may be removed by washing with deionized water and does not affect subsequent steps in western blotting. Ponceau S has low sensitivity, with a relatively low limit of detection (250 ng). Other protein stains include colloidal gold, India ink and Coomassie Brilliant Blue. However, these cannot be readily reversed with water and impede subsequent immunoprobing.
Coomassie Brilliant Blue may be used to stain the polyacrylamide gel to identify any proteins that have failed to transfer. Establishing if protein transfer is complete is important for downstream analysis. Therefore, it is essential to optimize the blotting process by adjusting the buffer composition and refining the transfer time.
Alternative Western Blotting Platforms
New techniques have been developed to increase productivity and flexibility. New platforms work to reduce analysis time, offer multiplexed detection and are high throughput by incorporating technologies such as capillary electrophoresis, microfluidics, and lab-on-a-chip microanalytical systems. These low-volume devices can process smaller amounts of protein, requiring smaller amounts of antibodies and other reagents and reducing the cost per assay. Faster processing makes high-throughput and multiplexed analysis possible.
Some new systems even demonstrate higher sensitivity in some cases compared to that achieved by conventional western blotting procedures. New techniques have been developed to facilitate automation, reducing the potential for user error observed in conventional western blotting, giving a standardized result. However, there are still improvements to be made before these devices become widespread.
Capillary Electrophoresis
Capillary electrophoresis separates proteins as they migrate through small capillaries filled with a polymer matrix that impedes their transit in a similar way to that of polyacrylamide gel.
Proteins elute from the capillaries onto a slow-moving membrane, allowing the protein size to be resolved. The proteins are then probed using conventional western blotting.
Microchip Capillary Electrophoresis
Microchip capillary electrophoresis (MCE) also uses capillary action to resolve proteins by size. A microchip is used in place of capillary tubes. The separated proteins are then captured on a membrane and probed by immunoassay.
In 2016, Jin et al. demonstrated higher sensitivity and better resolution with an MCE device in comparison to traditional western blotting.
The Herr group has further developed a device that miniaturized the whole western blotting process; a photoactive gel captures separated proteins by photoinduced cross-linking of the proteins to the gel, followed by in situ immunoprobing.
Single Cell Western Blotting
Single-cell western blotting is achieved using a multilayered microfluidic device, allowing cell-to-cell variations to be analyzed. Individual cells are placed in microwells, and lysis reagents are added via a “lid layer.”
A polyacrylamide gel base layer then separates the proteins using bi-directional electrophoresis. This technique has been demonstrated to achieve subcellular resolution and with a throughput of 103 cells in six hours.
Found Part 3 of our Western blotting guide useful? View part 4 here
References
- Blancher C. et al. (2001). SDS-PAGE and Western Blotting Techniques. Methods in Molecular Medicine. doi: 10.1385/1-59259-136-1:145.
- Jackson ImmunoResearch Laboratories Inc. (2017). Western Blotting Troubleshooting Guide! https://www.jacksonimmuno.com/secondary-antibody-resource/technical-tips/western-blot-trouble-shooting/
- GE Healthcare. (2011). Western Blotting Principles and Methods. https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma-Aldrich/General_Information/1/ge-western-blotting.pdf
- Bass J.J. et al. (2017). An Overview of Technical Considerations for Western Blotting Applications to Physiological Research. Scandinavian Journal of Medicine and Science in Sports. doi: 10.1111/sms.12702.
- Yamauchi K.A. et al. (2017). Subcellular Western Blotting of Single Cells. Microsystems and Nanoengineering. doi:10.1038/micronano.2016.79.
- Sanders B.J. et al. (2016). Recent Advances in Microscale Western Blotting. Analytical Methods. doi:10.1039/C6AY01947A.
- Mishra A. et al. (2017). Protein Purification and Analysis: Next Generation Western Blotting Techniques. Expect Review Proteomics. doi:10.1080/14789450.2017.1388167.

| Learn more: | Do more: |
|---|---|
| Colorimetric western blotting | Spectra Viewer |
| Chemiluminescence western blotting | Antibodies for signal enhancement |
| Fluorescent western blotting | |
Technical Resources | About us | Contact us | Bulk Service
Licenses | Conditions of Use | Privacy Policy | Ordering Information

FM 545248